Our long-term goal is to understand the molecular mechanism and regulation of human protein synthesis initiation on cellular and viral mRNAs.
Cellular mRNAs:
Cap-dependent mRNA recruitment to the ribosome requires the formation and maintenance of fundamental interactions between eukaryotic initiation factors (eIFs), the mRNA and the 40S subunit. Following mRNA entry into the decoding site, the 40S subunit locates the correct AUG codon by a scanning mechanism. Upon AUG recognition, the 60S subunit joins to form the 80S ribosome so that elongation can take place. This complicated mechanism requires a number of initiation components that possess a molecular mass roughly equal to that of the 40S subunit.
![]()
Cold Spring Harb Perspect Biol. 2019 Feb 1;11(2):a032706.
Cellular mRNA recruitment:
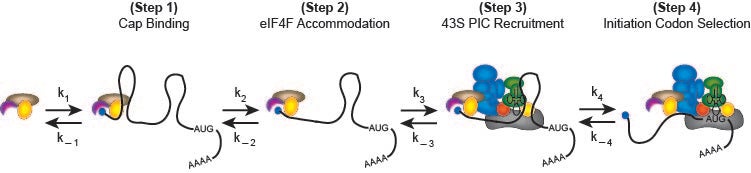
A possible kinetic pathway to describe the recruitment of cellular mRNAs to the human 40S ribosome (likely limiting relative to cytoplasmic mRNAs) is depicted in the following figure:
Biochimie. 2015 Jul;114:58-71
In step 1, the mRNA binds to the m7G cap-binding complex, eIF4F, which is composed of three proteins named eIF4E, eIF4G and eIF4A. eIF4E binds the m7G cap structure and the molecular scaffold eIF4G. The third component is the DEAD-box helicase, eIF4A, which provides the ATP-dependent helicase activity needed to unwind mRNA structure or remove or mRNA binding proteins to enable recruitment to the 40S subunit. In step 2, m7G cap proximal secondary structure is removed to enable full accommodation of the eIF4F complex. In step 3, the eIF4F–mRNA complex recruits the 43S pre-initiation complex (43S PIC; 40S subunit, eIF1, eIF1A, eIF2-GTP-Met-tRNAi and eIF3), possibly involving further helicase activity. Scanning of the 40S subunit downstream to the initiation codon occurs in step 4. Although not depicted, subsequent steps involve the docking of the 60S ribosomal subunit to form the 80S initiation complex at the initiation codon.
We anticipate that mRNAs are selected or rejected at the proposed checkpoints and that initiation factor modifications will alter the ability of different mRNAs to overcome checkpoints. Changing the overall rate of initiation (translation efficiency) will presumably be controlled by the rate at which individual checkpoints are overcome. Assays to monitor individual checkpoints are therefore needed to answer these questions on an mRNA-by-mRNA basis.
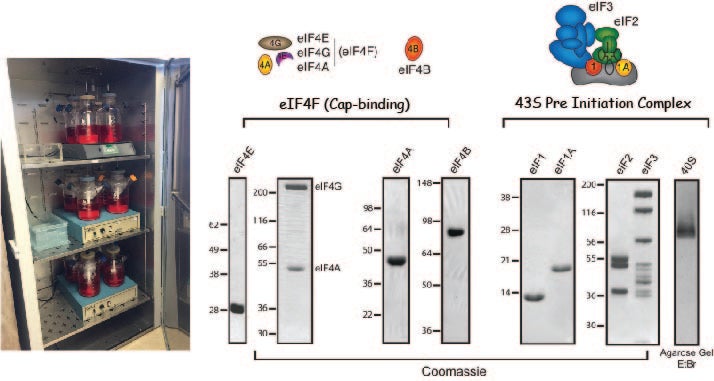
A reconstituted human initiation system: We have developed a reconstituted purified system to precisely monitor initiation factor activities and examine kinetic checkpoints in the human initiation pathway. Our system utilizes recombinant initiation factors where possible together with components purified from HeLa cell cytoplasm. Fluorescently modifying a number of initiation factors and mRNAs provides us with powerful signals to be exploited for bulk and single molecule assays.
Monitoring secondary structure remodeling in real time: To address how mRNA secondary structure can regulate kinetic checkpoints, we have generated a real-time unwinding assay to determine the rate of duplex unwinding by purified components (Nat Protoc. 2014 Jul;9(7):1645-61). This assay has identified eIF4B as a key component that couples the ATP hydrolysis cycle of eIF4A with duplex strand separation (J Mol Biol. 2011 Sep 30;412(4):674-87). It also enabled us to make the unexpected discovery that human eIF4E possesses a second function in translation initiation by strongly stimulating eIF4A helicase activity (Proc Natl Acad Sci U S A. 2013 Aug 13;110(33):13339-44). This unanticipated link between eIF4E and the helicase activity of eIF4A may help to explain why eIF4E over-expression promotes the translation of “structured” mRNAs during cellular transformation. We have also shown that this function of eIF4E also promotes Poliovirus translation in the absence of eIF4E cleavage (Proc Natl Acad Sci U S A. 2017 Sep 5;114(36):9611-9616).
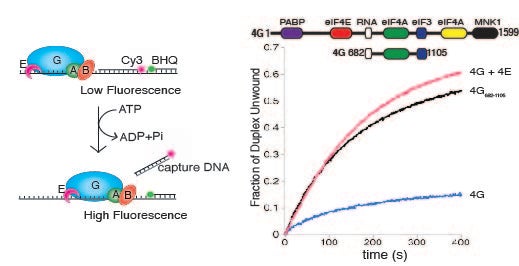
Proc. Natl. Acad. Sci. U. S. A. 2013 Aug 13:110(33);13339-44
In collaboration with Steven Block, we employed a single molecule assay to reveal that eIF4A becomes a processive helicase upon binding eIF4G and eIF4B (Science. 2015 Jun 26;348(6242):1486-8).
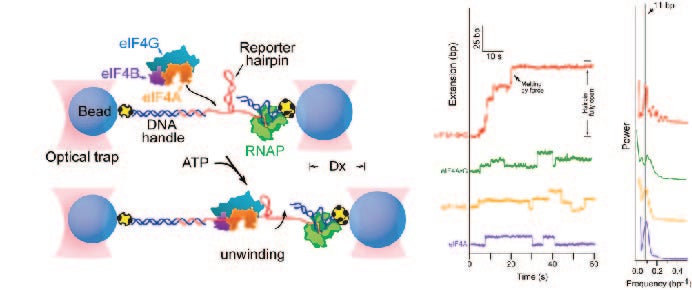
Science 2015 Jun 26:348(6242);1486-8.
Identifying interactions between components of the 43S preinitiation complex: The formation of the 43S PIC is an important intermediate in the initiation pathway. We have used fluorescence anisotropy to determine how the different components of the 43S PIC contribute to its stability. Our thermodynamic framework quantitatively defined how eIF2 and eIF3 function to stabilize the human 43S PIC (J Biol Chem. 2014 Nov 14;289(46):31827-36). This framework will help in our understanding of the emerging high-resolution structures of the 43S PIC from different organisms.
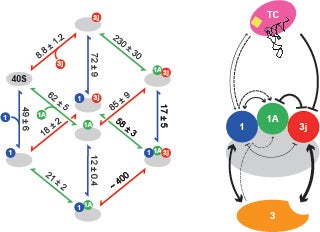
J. Biol. Chem. 2014 Nov 14:289(46):31827-36.
Recruitment of an mRNA to the 43S preinitiation complex: The direct interaction between eIF4G and eIF3 acts as a molecular bridge between the cap-binding complex and the 43S PIC. Using site-specific cross-linking, we have revealed that two distinct eIF3-binding domains exist in human eIF4G that bind to eIF3c, -d, and -e (J Biol Chem. 2013 Nov 15;288(46):32932-40). Binding to two non-core eIF3 subunits helps explain why the interaction between eIF4G and the 43S PIC may be different in yeast and mammals. Our current work is focused on generating a real-time fluorescent assay to monitor the recruitment of mRNA to the 43S PIC.
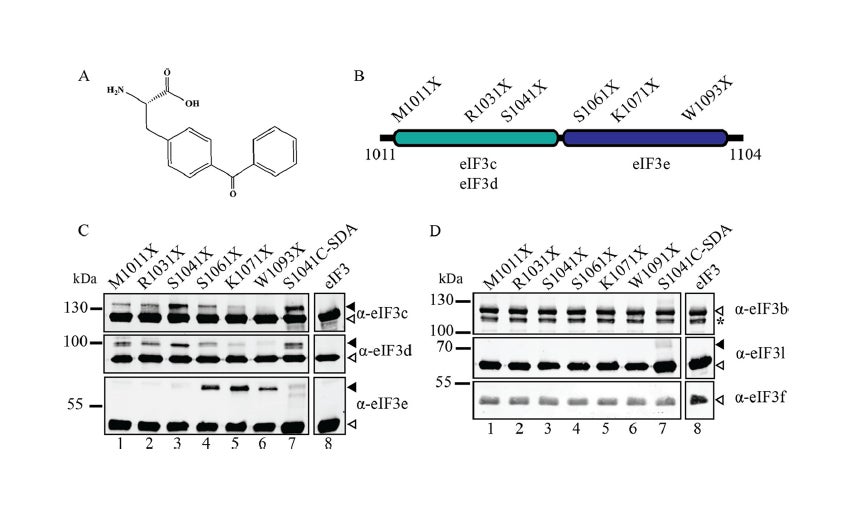
J. Biol. Chem. 2013 Nov 15:288(46):32932-40.
We have used a fluorescence-based anisotropy assay to quantitatively determine how initiation components coordinate their activities to reduce the affinity of eIF3j during the recruitment of mRNA to the 43S PIC. We showed that a full reduction in eIF3j affinity for the 43S PIC requires an ATP-dependent, but unwinding-independent, activity of eIF4A. This result suggests that in addition to its helicase activity, eIF4A uses the free energy of ATP binding and hydrolysis as a regulatory switch to control the conformation of the 43S PIC during mRNA recruitment. Our results define eIF4A as a universal initiation factor in cap-dependent translation initiation that functions beyond its role in RNA unwinding (Proc Natl Acad Sci U S A. 2017 Jun 13;114(24):6304-6309).
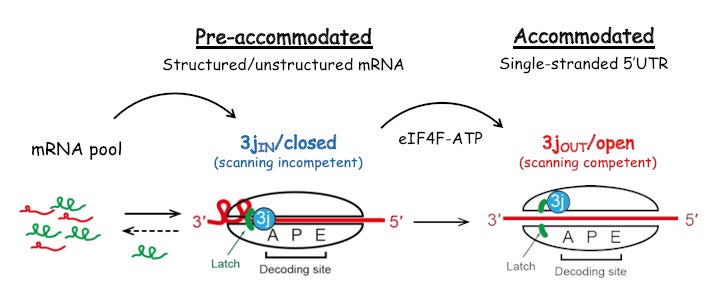
Proc. Natl. Acad. Sci. U. S. A. 2017 Jun 13:114(24):6304-6309).
Initiation factor modifications. Working with Julie Leary (UC Davis), we have been identifying the stoichiometries of post-translational modifications sites on eIF2, eIF3 and eIF4G (Int J Mol Sci. 2014 Jun 27;15(7):11523-38). We are currently investigating how specific signaling pathways converge to reprogram the translation machinery via changes in the phosphorylation of these initiation factors.
Viral mRNAs:
Many viral mRNAs recruit the ribosome using a cap-independent mechanism. One example is the hepatitis C virus, which uses a large RNA structure that forms an internal ribosome entry site (IRES). This structure is able to directly recruit the 40S subunit and promote initiation using a subset of initiation factors without the need for scanning.
Viral mRNA recruitment:
The HCV IRES consists of three highly conserved structural domains that interact specifically with the 40S subunit and the eIF3 complex. These interactions serve to position the 40S subunit directly at the translation start site in the viral mRNA, bypassing the need for the canonical scanning mechanism. Using the HCV IRES as a model, we are investigating the pathway of mRNA entry into the 40S subunit decoding site. Employing a site-directed RNA cleavage assay, we have provided a structural model for how eIF3j competes with mRNA for binding into the 40S subunit mRNA entry channel (Nat Struct Mol Biol. 2009 Apr;16(4):397-404). Our work indicates that in addition to a conformational change in the 40S subunit induced by the HCV IRES, the stability of the mRNA in the decoding site requires the recruitment of the eIF2-tRNAi complex to the 40S subunit. Our current work is focused on determining how different domains of the HCV IRES have evolved to functionally replace the different activities of canonical initiation factors for mRNA recruitment.
The Type 1 (e.g. poliovirus) and Type 2 (foot-and-mouth disease virus) picornavirus IRESes require eIF4G and eIF4A for their translation. We are interested in understanding how these IRESes use their RNA domains to promote mRNA recruitment to the 40S subunit to successfully compete with cellular mRNAs for translation.
Recruitment of poliovirus (PV) RNA to the human ribosome requires the coordinated interaction of the viral internal ribosome entry site (IRES) and several host cellular initiation factors and IRES trans-acting factors (ITAFs). Attenuated PV Sabin strains contain point mutations in the PV IRES domain V (dV) that inhibit viral translation. Remarkably, attenuation is most apparent in cells of the central nervous system, but the molecular basis to explain this is poorly understood. We used a fluorescence anisotropy assay to reveal that the Sabin mutants reduce the equilibrium dissociation constants of eIF4G and PTB to the PV IRES by up to 6-fold. Using the most inhibitory Sabin 3 mutant, we used a real-time fluorescence helicase assay to show that the apparent affinity of an active eIF4G/4A/4B helicase complex for the IRES is reduced by 2.5-fold. The Sabin 3 mutant did not alter the maximum rate of eIF4A-dependent helicase activity, suggesting that this mutant primarily reduces the affinity, rather than activity, of the unwinding complex. To confirm this affinity model of attenuation, we show that eIF4G overexpression in HeLa cells overcomes the attenuation of a Sabin 3 mutant PV-luciferase replicon. Our study provides a quantitative framework for understanding the mechanism of PV Sabin attenuation and provides an explanation for the previously observed cell type-specific translational attenuation (J. Biol. Chem. 2018 Oct 5;293(40):15471-15482).
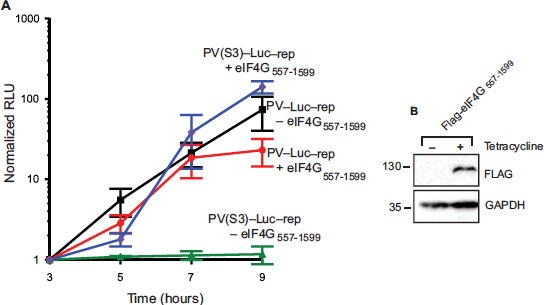
J. Biol. Chem. 2018 Oct 5;293(40):15471-15482.